

ˮ�еĿ�������Ⱦ��ȫ���Եĭh�����}����ʹ�ڵ͝����Ҳ������Ȼ���Bϵ�y����ش�Σ����Ŀǰ�_�l��������Ĥ���x�����オ�⡢���ۺ���������AOPs���ȶ�N̎������������ˮ�еĿ����ء�AOPs������x���Խ���ߌ����Զ��ܵ��V���Pע���c����OH·�ķ��D������ȣ��^�����}��AOPs���б������c���������L�İ�˥�ڡ����V���Ĺ���pH�������ߵ�����߀ԭ�λ����ˣ��漰�^�����}��ˮ�����^�������˿ƌW�ҵďV���Pע��
�ڌ��H�����У������^һ���ᣨPMS�������ֽ⾏���������Ҫ���������mȻ�ѽ��������S���������PMS����������ݗ�ա������ͼӟ�ȣ������ٴ����ļ���J��������Ч�ķ�����Ann���˜yԇ���S�ͬ�Ľ��٣��_�J��Co2+�ǻPMS����Ч�ķ��F���١�Ȼ������x�ӱ�횱��ι̵ع̶��ڿɻ��յĻ����ϣ����δ��ȫ���յĽ����x�ӿ��ܕ������ص���Ⱦ����С�F�غ͆�ԭ��λ�c�����Ȳ����ѱ�����AOPs����������ߴ�������ԭ������Ч�ʡ�Ȼ�����@Щ���Դ���ͨ���r���F�����칤ˇ���s����Ҫ���_������ĸߜ؟���^�̡�ͬ�r���@Щ����ͨ������{�״�С�������оۼ��A���@�����ؽ��ʹ����ԡ�����Ҫ���ǣ������ķ��x�ͻ�����Ȼ�o������Q�����������������Č��H���á���ˣ��Ƃ���и߱ȱ���e�����ܶȴ�λ�c�����Ժ߷����ԵĴ�ߴ磨����������Ȼ��һ���������


XAFS�Y���o���������ĽY���Pϵ���D2�@ʾ��Co�x�ӵľ����Y���������B������X�侀���ս�߅�Y����XANES���͔UչX�侀���վ����Y����EXAFS���V����7730��ӷ��ظ����ķ��ʾCo�ăr�B�Ԟ�+2����+3���ڈD2b�У������~׃�Q�UչXAFS�@ʾ��һ����1.5?�����ķ壬���w����Co-O��Co-N��һ���ӵ�ؕ�I���c�D�е�CoO��Co2O3�V��ȣ�P-PCaCo�V�е�2?��2.5?̎�ķ唵ֵ�������������]����������ڡ��@�C����Co�����÷�ɢ���cXPS��XRD�ĽY��һ�¡�Co:O��Co:N�ı����s��4��2�����Co�x����ȫ���ɰ�뿻����ϣ�Co:N���ʑ�����Co:O���ʡ�ͨ�^�����I�L�ͽj�ϔ����_����Co�Ƀɂ��ɰ�뿵�ԭ�Ӻ��Ă�ˮ������λ���D2d�f����Co����λ�Y�����@Ҳ�õ���DFTӋ��Y����֧�֡�
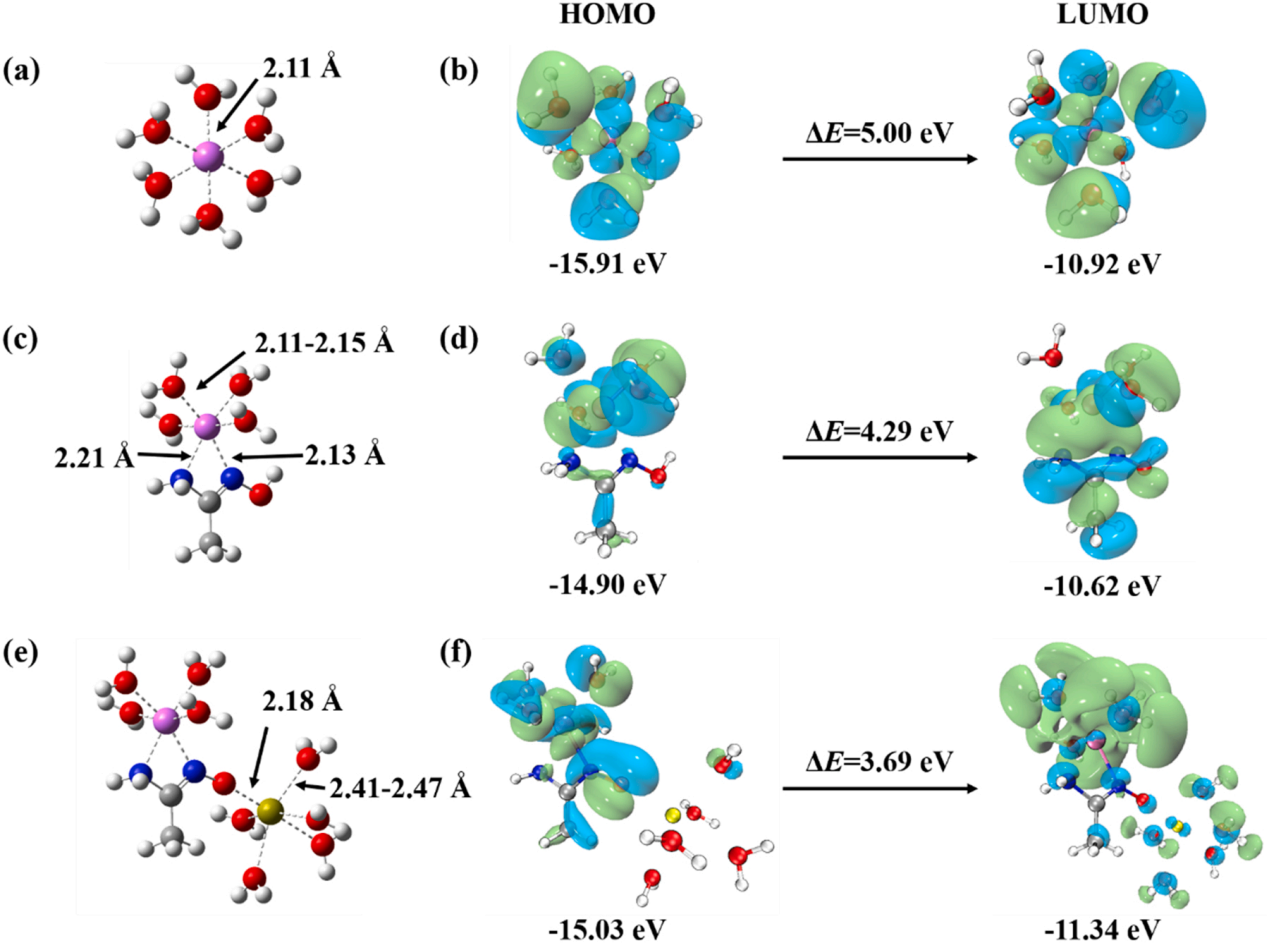
�D3.(a) [Co(H2O)6]2+��(c) AO-Co��(e) AO-CaCo������ĽY����(b) [Co(H2O)6]2+��(d) AO-Co ��(f) AO-CaCo��ǰ��܉����HOMO-LUMO gap��

�D4. (a)AO-CaCo���o늄ݷֲ���(b)AO-CaCo���o늄ݷ����A���ӱ��棻(c) [Co(H2O)6]2+��(d) AO-Co��(e) AO-CaCo��PMS�ķ������g�w
�������ӵı���늺ɱ��������Mһ���_�J�}�x�ӌ����^�̵Ĵ��M����D4��ʾ�� AO-CaCo���o늄ݣ�ESP���yӋ�������������ӱ�����зdz��ߵ���늺ɣ�����ؓ늵�PMS�x�Ӿ��Џ��ҵ������������ڛ]��ؓ늺Ʌ^����С늺Ʌ^��ķ����Եͣ����@���^���Л]�����x������o늄݅^��ļ�����D4b��ʾ���^������o늄��cλ��Co���棬ֵ��10.29 eV���@��ζ�����ǻ�����ߵ�λ�á��@�ٴ��f����Ca2+��Co2+֮�g��늺��D�ơ�����ESP�Y����Ӌ���˷������g�w����D4c-e��ʾ��PMS��Co2+�ĽY�Ͼ��к��Ą����W����������AO-CaCo���������@����Ȼ����AO-Co�Ħ�E����[Co(H2O)6]2+���@�����������ɰ�뿻��F��ؓ늺����¡�������Ca2+���@���ӄ���ȫ�����D���C���������@Щ�Y��������P-PCaCo������Co2+������ˮƽ��늺ɣ��Ķ����M��PMS�ĽY�Ϻʹ��ֽ⡣����P-PCaCo��˸ߵ���늺ɲ�����������늵��}���ĭh�ص��ЙC��Ⱦ����PMS�����ѽ�����^��·���ֽ⣬�γɸ��NAOPs��Ȼ���c�ЙC��Ⱦ�ﷴ����
ͨ�^ݗ�����l��֦�ۺ��@�N�����������˟o������������ɷN�����x�ӣ��Ķ��_�l��һ�N���͵�PMS������ԓ���������Ƃ�ͻ��գ������F���O�ߵĴ����ܡ�PE/PET�o�������ײ��H�ṩ�˷����ԡ����؏�ʹ���Ժ߱ȱ���e��߀ͨ�^��Һ�еĶ��ο��g���x�����˴�λ�c��ԭ�ӷ�ɢ�ȡ�Ca2+������������ͨ�^��ֹCo�ľۼ�����������o늄݁�f�����^�̡����⣬Ca2+�ڹ�ܗ�Y���г䮔����������������w��Ч�ʲ������˴������܉���P-PCaCo�����ڲ�ͬ�l���¿�����8��犃Ƚ���TC��չʾ����ɿ��ԺͶ���ԡ��Y�����������ɻ�������˻��W�Y���Ϳ��ܵĴ��^�̡���ՓӋ���Mһ������˽Y���ߴ����ܡ��@��о����l��̽����ƺ��������Ƃ��Ч���õĴ������阋��ͬ�r���д���;����Y���Ĵ����ṩ��һ�N��˼·��
ԭ��朽ӣ�http://dx.doi.org/10.1016/j.apcatb.2023.122698
- ���ƴ�����ܡ����W�������֡��B��/������ƽ JACS�����w���M���~��DArP�ϳɸ������ЙC����x����ӌ��w 2026-03-09
- �L��������ꐌW˼/���@�F� Angew���������Ч���{�ص��p�����F�������h�������c�h�������ɿع���-�ϳɸ߷������������� 2026-03-05
- �ӹ����eԪ���� Macromolecules���h����˨���ЙC��·��˹��A�� - ������ϡጵĸ�Ч�ЙC���������CO2�ͭh�����鹲�� 2026-02-26
- �Ͽƴ�����T���F� Angew��������w���ʵ��p�������������ۺ������w����������ȫ�ۺ���̫���늳� 2023-07-21
- �Ͽƴ�����ڈF� AFM����ԭ��ȡ��ͬ�r�������w���ʺ͓��sЧ�ʌ��F������n�͟�늲��� 2023-03-21
- �A��������W�w�����ڡ�Adv. Sci.��������ֱ������ - �ۯB�Ϳ��g��ܗ��ӂ�ݔ���� 2022-04-01
- ���ִ�W���Ŀƽ��ڈF� Macromolecules���ۺ����w�S���������c�η������W�О�Ķ����P 2025-10-15
