研究背景
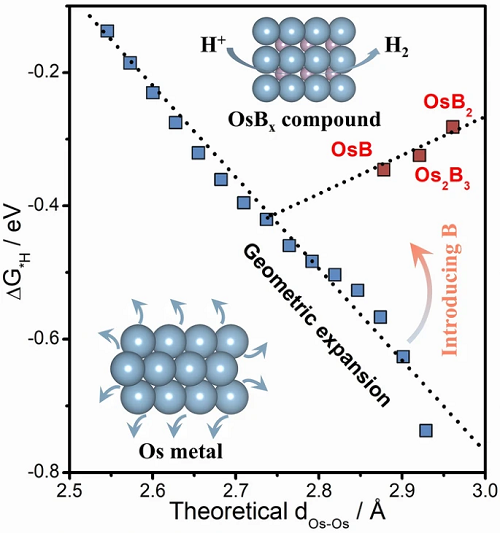
在原子尺度上精確調整金屬活性中心間距對提高催化活性和加深對催化機理的認識具有重要意義�����,但仍然充滿挑戰。對此,武漢理工大學木士教授課題組利用輕原子稀釋活性金屬位點密度的方法實現了金屬原子間距(dM-M)的精確可調,同時還發現了一種有別于常規的氫吸附模式。以活性金屬鋨(Os)和作為填隙原子的硼(B)為例��,在OsBx金屬間化合物結構中���,如果逐漸增加B的含量��,將會伴隨著Os金屬位點密度的減少����,Os的dOs-Os將逐漸從2.73增加到2.96 A����。然而,隨著dOs-Os增加���,氫的吸附-距離關系通過d帶的下移而出現反轉,打破了傳統的認知,從而優化了催化過程中氫在電極表面的吸附和H2O的解離�����,最終使析氫反應(HER)活性幾乎呈線性增加�����。在堿性介質中�����,具有最大dOs-Os值(2.96 A)的OsB2表現出最佳的HER活性(8 mV @ 10 mA cm-2),而且還抑制了O的吸附,從而提高了催化劑的穩定性。這種新型的原子級催化位點距離調制策略和氫的吸附-距離反轉關系的發現為高效催化劑的設計提供了新的見解���。
Tuning Active Metal Atomic Spacing by Filling of Light Atoms and Resulting Reversed Hydrogen Adsorption?Distance Relationship for Efficient Catalysis
Ding Chen*, Ruihu Lu*, Ruohan Yu, Hongyu Zhao, Dulan Wu, Youtao Yao, Kesong Yu, Jiawei Zhu, Pengxia Ji, Zonghua Pu, Zongkui Kou, Jun Yu, Jinsong Wu, Shichun Mu*
Nano-Micro Letters (2023)15: 168
https://doi.org/10.1007/s40820-023-01142-1
本文亮點
1. 密度泛函理論(DFT)計算表明,間隙B原子可以調整主金屬Os的原子間距,并表現出反轉的氫吸附-距離關系�。
2. 建立了活性Os原子間距與催化活性之間的構效關系�。
3. 在構建的多個OsBx物相中�,具有最大B:O原子比的OsB2(dOs-Os=2.96 A)在堿性介質中表現出最佳的HER活性(8 mV @ 10 mA cm-2)和良好的電化學穩定性。
內容簡介
利用間隙填充策略實現活性原子間距的精準調控及構效關系的解讀具有重要科學意義,但該方面一直是研究的空白。武漢理工大學木士春課題組通過密度泛函理論(DFT)計算,首次證實了小半徑和低電負性的硼(B)原子能夠完美地平衡金屬鋨(Os)幾何膨脹過程中的應力變化和電子轉移����;伴隨著金屬間化合物OsBx(x=1����,1.5����,2)中B間隙原子數量的增加,Os金屬位點密度減少,Os原子間距(dOs-Os)從2.73逐漸增加到2.96 A,并獲得了反轉的氫吸附-距離關系,即間隙B原子填充在增加Os原子間距(dOs-Os)的同時亦削弱了Os對氫(H)的強吸附���。與傳統的表面修飾和摻雜策略不同,強的主客體電子相互作用和新化學鍵的形成進一步增強了催化劑的活性和穩定性。此外,通過研究還構建了OsBx的結構-活性關系���,即活性Os原子間距隨著B的逐漸填充而增大����,導致H結合和H2O解離能壘減小�;同時��,增強的Os-B配位效應抑制了Os的失活和溶解,實現了催化劑活性和穩定性的同步提高�。
圖文導讀
I 密度泛函理論(DFT)計算
理論計算結果如圖1所示��。圖1a表明,OsBx金屬間化合物隨著B含量的增加����,Os原子間距離dOs-Os從2.73逐漸增加到2.96 A��,進一步導致空位(hollow site)的Os-H鍵長增加;圖1b表明����,與傳統認識相反���,具有小半徑和低電負性的間隙B可以實現反轉的氫吸附-距離關系�����;從圖1c可看出,幾何膨脹通常會導致Os-Os相互作用減弱���,導致d帶中心(εd)升高,吸附能力增強����;而B間隙原子可以通過降低d帶實現氫吸附-距離關系的反轉��。上述理論分析揭示了通過填充間隙原子B來梯度分散Os活性位點及提高HER活性是可行的,為后續數類似催化劑的合成和構效關系的確定指明了方向�����。

圖1. 理論計算��。(a)原子間距調制,紫色、藍色和白色的球分別代表B����、Os和H原子���;(b) ΔG*H隨Os-Os鍵長增加的變化����,箭頭表示引入B的效果;(c)B引入引起的d帶位移示意圖��。
II 結構表征
如圖2a表示��,通過控制退火溫度��,熱解得到三種金屬間硼化物;圖2b的XRD結果表明���,在700℃、800℃和900℃下得到的產物分別為六方晶相OsB (OsB- H)和Os2B3 (Os2B3-H)�,正交晶相OsB2 (OsB2-O)����;圖2c顯示了Os�、OsB、Os2B3和OsB2四個典型的Os L3邊EXAFS譜圖��,表明填充間隙B原子誘導了新的Os-B鍵的形成以及dOs-Os逐漸增加; 圖2d進一步描述了實驗和理論得到的dOs-Os之間的線性擬合結果���,證實了間隙B填充引起的Os原子的逐漸分散���;圖2e-h展示了沿Os [100]�����,OsB-H [010],Os2B3-H[001]和OsB2-O[101]帶軸的高分辨率晶格原子圖像和相應的快速傅里葉變換(FFT)模式,與理論的晶體結構和原子排列非常吻合�。以上結果充分證明了一系列有序金屬間硼化物的合成��,并實現了Os金屬原子的可控分散。
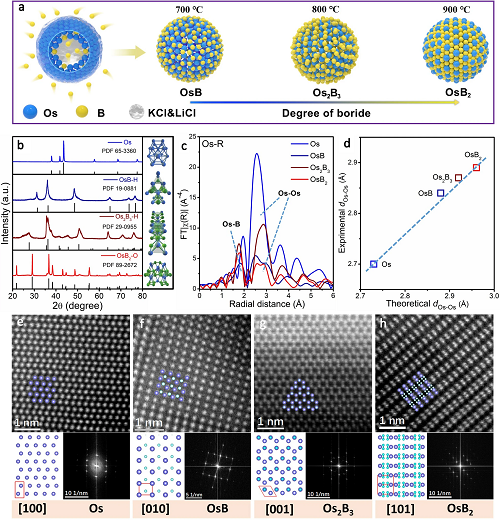
圖2. 結構表征。(a)制備OsBx的示意圖; (b)所有合成材料的XRD圖譜; (c)Os、OsB����、Os2B3和OsB2的L3 邊EXAFS光譜; (d)理論與實驗Os-Os距離的線性關系�����;(e-h)高分辨率HAADF-STEM圖像及其相應的晶體結構和FFT圖,其中Os原子呈藍紫色,B原子呈藍白色�����。
III 析氫反應(HER)活性
圖3a-b表明OsBx在1 M KOH中的HER活性和動力學優于Os和商用Pt/C�����;圖3c表示在過電位為50 mV時����,OsB?具有最高的轉換頻率(TOF)���,分別是Os2B3和OsB的4.0倍和2.4倍�;圖3d顯示了酸性介質中HER性能與ΔG*H之間良好的線性擬合關系,表明Os轉化為OsB2導致的*H吸附減弱是促進HER活性的主要原因之一;圖3e表示在Os����、OsB、Os2B3和OsB2的Os位點上�����,H2O解離是速率決定步驟����,限制了HER過程;圖3f進一步對50 mA cm-2下的能壘和堿性HER活性進行擬合�,良好的線性關系證明間隙B原子加速了H2O解離���,從而提高了堿性HER活性��;綜上所述����,B原子的引入減弱了*H吸附����,加速了H2O解離。圖3g比較了OsBx與最近報道的鉑族金屬電催化劑在1 M KOH條件下的HER性能,相比于大多數的Pt簇金屬基催化劑����,OsB2具有更好的HER活性�。
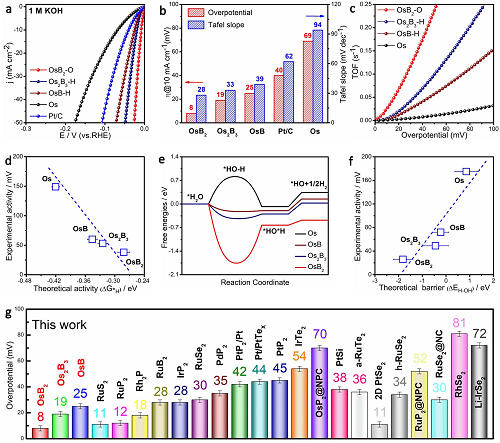
圖3. HER活性����。(a)HER極化曲線;(b)在10 mA cm-2下對應過電位和Tafel斜率;(c)TOF與Os和OsBx過電位之間的關系�;(d)酸性條件下理論與實際活性的相關性��;(e)堿性HER的自由能圖;(f)堿性環境下H-OH分裂能壘與實際活性的關系;(g)比較OsBx與最近報道的鉑族金屬電催化劑在10 mA cm-2�����、1 M KOH條件下的HER性能�����。
IV 活性提高機理
圖4a-b表明成功引入了間隙B;圖4c的小波變換可視化了Os-B路徑�;圖4d-e通過對比1500~1700 cm-1的原位拉曼光譜的寬峰���,可以進一步證明OsB2相對于Os對H2O的吸附也明顯減弱�;圖4f通過UPS探測Os和OsBx催化劑的占據電子態來了解B-O相互作用對氫吸附能力的影響����,表明B和Os原子之間的p-d雜化導致Os的d帶下移,使OsBx的εd發生負移���;圖4g顯示了實驗和理論εd之間良好的線性關系,進一步證明了εd是通過B-Os相互作用而下降的�;圖4h表明εd的下降減弱了Os 5d和H 1s之間的反應性��,導致*H吸附較弱;圖4i表示d帶理論預測的這一趨勢與計算得到的*H吸附也很吻合��。

圖4.活性提高機理�。(a)對應產物B 1s的XPS譜圖;(b)OsB?的L?邊EXAFS擬合曲線��;(c)EXAFS信號的WT�����;(d-e)Os和OsB2的原位拉曼分析����;(f)Os���、OsB、Os2B3和OsB2相對于費米能級的UPS價帶譜����;(g)實驗與理論εd之間的校正;(h)*H s軌道與Os 5d軌道的相互作用;(i)理論εd��、ΔG*H與HER活性的關系���。
V 穩定性提高機理
最后���,研究人員還探索了OsBx穩定性提高的機理��。圖5a表示催化劑在電解質中的活性降解可能是由于金屬氧化態的形成導致了兩條活性失效路徑�����;圖5b表示OsB、Os2B3和OsB2活性中心的Os原子表面εd分別為-1.24��、-1.51和-1.91 eV�。根據d波段理論����,通過*O吸附而使εd上移會使反應性增強���,從而導致*H吸附增強和HER性能下降��,因此豐富的Os-B鍵抑制了O的吸附���;圖5c-d分別為不同加速循環后催化劑的過電位與εd的關系�����,以及電催化后OsB、Os2B3和OsB2溶解在電解液中Os的濃度�。這些結果中OsB?的更優表現進一步證實了金屬間硼化物結構越復雜,配位效應越強,在HER過程中抗氧化中毒和溶解穩定性越好。圖5e進一步表示OsB2在酸性和堿性介質中耐久性試驗(100 h)中都能保持穩定的工作電流�。
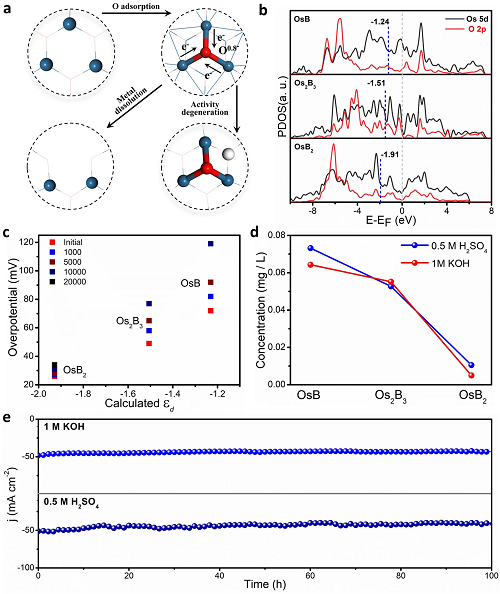
圖5. 穩定性提高機理�。(a)強O吸附誘導催化活性退化的兩種可能機制�;(b)相關氧原子和吸附氧的O2p軌道和O5d軌道的PDOS計算;(c)在1 M KOH中不同加速循環后催化劑的過電位與氧的關系;(d)電催化后OsB�、Os2B3和OsB2溶解的電解質中Os的濃度���;(e)OsB2在1M KOH和0.5 M H2SO4條件下的耐久性�����。
作者簡介

陳釘
本文第一作者
武漢理工大學 博士研究生
▍主要研究領域
催化劑的設計合成與催化機制分析。

木士春
本文通訊作者
武漢理工大學 教授
▍主要研究領域
(1)燃料電池催化劑;(2)電解水產氫催化劑。
▍個人簡介
武漢理工大學首席教授����,博士生導師�����。長期致力于氫能源催化材料研究。以第一作者或通訊作者在Nat. Commun.、Adv. Mater.�����、J. Am. Chem. Soc.���、Angew. Chem. Int. Ed.����、Energy Environ. Sci.等國內外期刊上發表280余篇高質量學術論文。
▍Email:msc@whut.edu.cn
撰稿:原文作者
編輯:《納微快報(英文)》編輯部