Energist 能源學人 2023-02-09 07:44 發表于廣東
【研究背景】傳統鋰離子電池低的能量密度和壽命已難以滿足日益增長的電驅動設備需求。具有超高比容量和低氧化還原電位的金屬鋰被視為最具潛力的下一代高比能二次電池的負極材料,但鋰金屬電池在實際應用中會持續受到鋰枝晶生長和體積膨脹的影響,導致不穩定的固體-電解質間相層(SEI)產生和庫侖效率的降低(CE)。同時,隨著電池長時間循環,鋰枝晶不斷生長并最終積累形成大量“死鋰”,還會帶來安全隱患。為了解決上述問題,研究者們已經采取了諸多改進策略。其中一個最為普遍的方法是通過修飾親鋰位點改善傳統集流體的疏鋰性。但目前很少有研究關注合金中親鋰金屬位點在脫合金化后的失效機制以及循環后成核位點的可持續性問題。
【工作介紹】近日,武漢理工大學木士春教授與何大平教授合作報道了一種獨特的“三明治”鋰金屬復合負極,其中納米級鋅(Zn)金屬被均勻地限制在氧化石墨烯(GO)和銅(Cu)箔之間。在這種獨特的結構中,親鋰的納米Zn顆粒在中間層作為鋰成核種子,有利于鋰的優先沉積并形成LiZn合金。理論和實驗研究結果表明,GO層作為人工SEI界面不僅為Li+的快速傳輸提供了通道,同時對Zn2+有很強的吸附性能,從而保證了成核晶種在長期循環過程中的穩定性。因此,與純合金負極和石墨烯保護的無合金負極相比,基于GO保護的復合合金負極具有更小的過電位和更優異的循環穩定性。其半電池可在1 mA cm-2下穩定循環200周,庫侖效率達98%以上;對稱電池在1 mA cm-2下可延長工作壽命至600h以上,且極化較低;基于該負極可進一步與商用LiFePO4正極匹配成全電池(11.5 mg cm-2)在5C的高倍率下保持90 mAh g-1的高比容量,而且在1C電流密度下循環100周后仍有81.1%的高容量保持率。這項工作為提高親鋰晶種的可持續性以高效儲存鋰提供了新的見解。該文章相關工作以題為“Stabilizing nucleation seeds in Li metal anode via ion-selective graphene oxide interfaces”發表在Energy storage material上。馬菁菁,楊金龍為本文共同第一作者。
【內容表述】本項研究設計出獨特的“三明治”鋰金屬負極,并研究了親鋰位點在脫合金化過程中的演變過程,隨后證明了氧化石墨烯(GO)在對鋅(Zn)晶種的穩定性起到了關鍵作用,有利于形成穩定的鋰金屬沉積-脫出。通過DFT計算提出GO對Zn2+有較強的吸附作用,據此推測GO可能會降低因LiZn合金出現Zn2+脫溶現象所帶來的不利影響。進一步實驗研究表明:1)在脫合金化過程中,Zn2+易從LiZn合金中進入電解液;2)GO對Zn2+有錨定作用,從而提高了Zn作為沉積位點的穩定性;3)同時,GO作為高鋰離子電導率的人工界面,可有效促進Li+的擴散,證實了理論預測結果。因此,本文發現,具有離子選擇性的GO作為人工保護界面有助于穩定親鋰Zn層,可有效抑制鋰枝晶生長。
1. 第一性原理模擬計算與實驗設計  圖1.(a)DFT模擬計算與統計與GO中不同官能團(P-G、C-O-G、C-OH-G、C-V、COG-V、COH-V)的18個不同位點對Zn2+、Li+的結合能大小。(b) Li+在 GO表面和層間遷移路徑的結合能和遷移能及示意圖(插圖)。(c) GO-Zn/Cu三層結構電極的設計及其作用機理。
圖1.(a)DFT模擬計算與統計與GO中不同官能團(P-G、C-O-G、C-OH-G、C-V、COG-V、COH-V)的18個不同位點對Zn2+、Li+的結合能大小。(b) Li+在 GO表面和層間遷移路徑的結合能和遷移能及示意圖(插圖)。(c) GO-Zn/Cu三層結構電極的設計及其作用機理。
循環過程中,Li+優先沉積在降低成核勢壘的親鋰Zn位點處,并形成Li/LiZn層。然而,當Zn/Cu作為電池負極進行脫鋰過程時會發生Zn2+的脫溶,從而影響親鋰層的穩定性。DFT計算發現GO中不同官能團所有相關位點對Zn的結合能均比對鋰的結合能更強(圖1a),說明Zn可以被GO錨定。因此,GO可作為人工界面保護層提高GO-Zn/Cu電極中親鋰層的可持續性。此外, Li+在GO表面遷移能壘較低,且形成原生SEI組分,有利于Li+在表面遷移至缺陷處,再通過缺陷(Void-i, ii, iii)穿透石墨烯(圖1b)。同時,GO具有很高的柔性和機械性能,有利于維持人工SEI的完整性。DFT模擬結果證實了GO作為界面材料具有保護負極和抑制鋰枝晶的先進特性。(圖1c)
2. GO-Zn/Cu合成過程中結構演變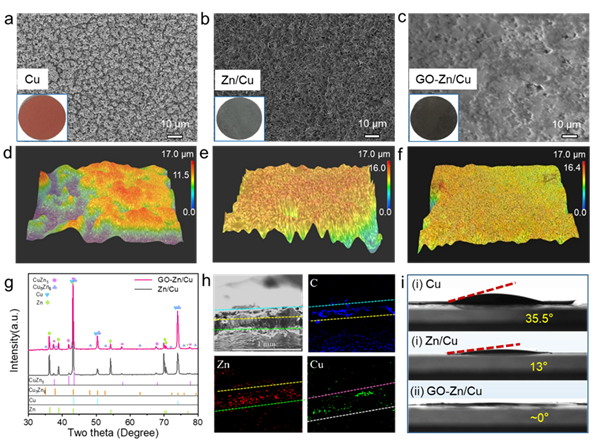 圖2.(a-c) GO-Zn/Cu、Zn/Cu、GO/Cu和純Cu基底的SEM和數字圖像(插圖),以及(d-f) Cu、Zn/Cu和GO-Zn/Cu的3D超景深圖。(g) GO-Zn/Cu和Zn/Cu電極的XRD譜圖。(h) GO-Zn/Cu電極的元素分析。(i)電解液與Cu、Zn/Cu和GO-Zn/Cu電極的接觸角大小。
圖2.(a-c) GO-Zn/Cu、Zn/Cu、GO/Cu和純Cu基底的SEM和數字圖像(插圖),以及(d-f) Cu、Zn/Cu和GO-Zn/Cu的3D超景深圖。(g) GO-Zn/Cu和Zn/Cu電極的XRD譜圖。(h) GO-Zn/Cu電極的元素分析。(i)電解液與Cu、Zn/Cu和GO-Zn/Cu電極的接觸角大小。
SEM和對應的光學照片(插圖)證實了GO-Zn/Cu電極從粗糙的Cu表面(圖2a),到納米鋅結構(圖2b),最后形成光滑的復合基底(圖2c)的演變過程。3D超景深顯微圖像進一步驗證此三維復合負極的均勻平整結構變化 (圖2d, e, f)。表面平整的基底有利于鋰均勻沉積,三明治結構中的納米Zn為電沉積鋰提供了親鋰位點,從而有效促進Li與Zn之間的反應,并形成高比容量負極材料Li/LiZn。同時,Zn/Cu電極的XRD圖譜顯示了CuZn合金相的存在(圖2g),說明加強電沉積后的Zn層與Cu箔之間存在緊密接觸,有利于降低界面電阻和解決了循環過程中活性物質脫落導致的電池循環穩定性問題。EDS結果顯示了部分Zn和Cu的界面發生融合,其中碳源來自GO(圖2h)。此外,GO的官能團和缺陷使GO-Zn/Cu電極與其他基底相比的潤濕性更好(圖2i),保證了電解質與基底之間更好的接觸,更加說明石墨烯是最佳的人工SEI材料之一。
3. 半電池電化學性能 圖3.GO-Zn/Cu、Zn/Cu和純Cu基底上鋰沉積,脫出行為(a) 比較電池之間的成核過電位。(b)不同基底作為工作電極組裝成半電池在50周循環后得到的擬合EIS譜和等效電路圖。(c)不同基底相對應的阻抗擬合數據(Rsei、Rct)。(d, e) 不同基底上在電流密度分別為(d) 0.5 mA cm-2、(e) 1.0 mA cm-2下的庫侖效率和(f) 第65圈充放電曲線。(g) 比較對稱電池Li@Cu、Li@Zn/Cu和Li@GO-Zn/Cu電極在電流密度為1 mA cm-2下的循環穩定性以及(h)不同倍率下的電壓變化情況。
圖3.GO-Zn/Cu、Zn/Cu和純Cu基底上鋰沉積,脫出行為(a) 比較電池之間的成核過電位。(b)不同基底作為工作電極組裝成半電池在50周循環后得到的擬合EIS譜和等效電路圖。(c)不同基底相對應的阻抗擬合數據(Rsei、Rct)。(d, e) 不同基底上在電流密度分別為(d) 0.5 mA cm-2、(e) 1.0 mA cm-2下的庫侖效率和(f) 第65圈充放電曲線。(g) 比較對稱電池Li@Cu、Li@Zn/Cu和Li@GO-Zn/Cu電極在電流密度為1 mA cm-2下的循環穩定性以及(h)不同倍率下的電壓變化情況。
GO-Zn/Cu電極在電流密度為0.2和1 mA cm-2時,金屬鋰沉積的成核過電位分別為9.3和22.0 mV,與其他電極相比成核過電位最低(圖3a)。該結果證實GO和Zn的協同作用降低了鋰成核勢壘。EIS測試表明,在多次循環后,GO-Zn/Cu基底優化了界面動力學,并推動了鋰離子的快速遷移(圖3b,c)。CE測試展示了GO-Zn/Cu||Li半電池在不同電流密度(0.5~2 mA cm-2)和容量(0.5~4 mAh cm-2)下的循環穩定性均明顯優于其他基底組裝成的電極 (圖3d-f)。ICP-MS測試進一步驗證GO-Zn/Cu電極中Zn2+會從LiZn合金中遷移出,且石墨烯界面層可有效地阻止這種現象。這與DFT計算結果一致,證實了GO作為鋰金屬負極界面層在保護活性位點和提高電池循環穩定性方面確實起到重要作用。進一步分析鍍鋰/脫鋰的過電位,對稱電池Li@GO-Zn/Cu產生20 mV的最低過電位,并可循環620 h(圖3g)。此外,Li@GO-Zn/Cu||Li@GO-Zn/Cu在不同倍率下表現出良好的循環穩定性和可逆性(圖3h),表明此復合結構能有效穩定鋰金屬負極。
4. 鋰沉積、脫出過程中電極截面結構變化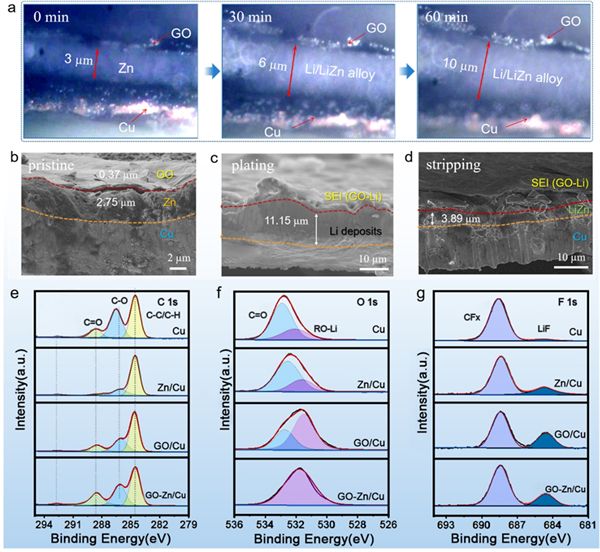 圖4.(a)在 1 mA cm-2電流密度下鋰沉積隨時間變化的原位光學顯微圖像。(b-d) GO-Zn/Cu電極的截面SEM圖像:(b)初始電極,(c)沉積1 mAh cm-2后的電極,(d)脫出1 mAh cm-2后的電極。在循環幾圈后Cu、GO/Cu、Zn/Gu和GO-Zn/Cu基底的XPS譜(e) C 1s, (f) O 1s, (g) f 1s。
圖4.(a)在 1 mA cm-2電流密度下鋰沉積隨時間變化的原位光學顯微圖像。(b-d) GO-Zn/Cu電極的截面SEM圖像:(b)初始電極,(c)沉積1 mAh cm-2后的電極,(d)脫出1 mAh cm-2后的電極。在循環幾圈后Cu、GO/Cu、Zn/Gu和GO-Zn/Cu基底的XPS譜(e) C 1s, (f) O 1s, (g) f 1s。
通過原位光學顯微圖像可以直接觀察到,隨著沉積時間的變化,鋰優先沉積在GO界面下的中間鋅層,且厚度逐漸增加(圖4a)。圖4b-d的非原位SEM圖像觀察結果與原位光學顯微圖像觀測結果一致。此外,采用高分辨率XPS光譜分析了4個電極的表面化學成分(圖4e-g)。結果表明,在循環過程中,醚基電解液分解,且在GO表面形成致密的原生SEI層,并保持完整性,能穩定加速離子傳輸。
5. 鋰沉積、脫出過程中電極表面結構變化 圖5.(a, e, i) Cu, (b, f, j) GO/Cu, (c, g, k) Zn/Cu和(d, h, l) GO-Zn/Cu基底在1mA cm-2電流密度和1.0 mAh cm-2容量下的第1次鋰沉積、剝離和第50次剝離后的SEM圖像。
圖5.(a, e, i) Cu, (b, f, j) GO/Cu, (c, g, k) Zn/Cu和(d, h, l) GO-Zn/Cu基底在1mA cm-2電流密度和1.0 mAh cm-2容量下的第1次鋰沉積、剝離和第50次剝離后的SEM圖像。
研究了四種電極在經歷不同循環和不同周期下的表面結構。第一次鋰沉積過程后,在Cu(圖5a)、GO/Cu(圖5b)、Zn/Cu(圖5c)電極上分別都形成了不同程度大小的枝晶,而GO-Zn /Cu復合電極中所形成的表面結構更加平滑均勻(圖5d)。圖5e-h顯示了循環第一次鋰脫出時,不同于粗糙的Cu(圖5e)、GO/Cu(圖5f)、Zn/Cu(圖5g)的電極表面,GO-Zn/Cu復合電極中仍保持Zn納米粒子,且均勻地附著在石墨烯界面層下(圖5h)。這意味著需要界面層與親鋰層的協同作用才能有效抑制枝晶生長和“死鋰”。再進一步對50次剝離后四種電極的表面結構進行了深入的對比分析。在Cu和GO/Cu基底上可以發現積累大量的“死鋰” (圖5i-j);對于Zn/Cu基底,由于在鋰脫出過程中存在晶種Zn部分脫溶情況,于是在Zn層的孔洞和裂紋附近產生了不均勻的鋰沉積和枝晶生長(圖5k);然而,GO-Zn/Cu復合電極在50次剝離后表面仍保持光滑平整,完全沒有觀察到鋰枝晶(圖5l)。基于上述結果,作者提出該類型中界面與親鋰層的協同效應可以完全抑制GO-Zn/Cu基底上枝晶和“死鋰”的產生。
6. 全電池電化學性能測試 圖6.(a) LFP||Li@GO-Zn/Cu全電池配比示意圖。(b, c)初始時(b)和一定角度彎折后(c)柔性軟包全電池供電發光二極管的照片。(d) LFP||Li@GO-Zn/Cu全電池對應的充放電電壓-容量分布。(e) LFP//Li@Cu、LFP//Li@GO/Cu、LFP//Li@Zn/Cu和LFP//Li@GO-Zn/Cu全電池在0.1、0.2、0.5、1、2和5C的不同倍率性能和(f)相應的極化電壓。(g)四種全電池在1C電流密度下的長循環性能。
圖6.(a) LFP||Li@GO-Zn/Cu全電池配比示意圖。(b, c)初始時(b)和一定角度彎折后(c)柔性軟包全電池供電發光二極管的照片。(d) LFP||Li@GO-Zn/Cu全電池對應的充放電電壓-容量分布。(e) LFP//Li@Cu、LFP//Li@GO/Cu、LFP//Li@Zn/Cu和LFP//Li@GO-Zn/Cu全電池在0.1、0.2、0.5、1、2和5C的不同倍率性能和(f)相應的極化電壓。(g)四種全電池在1C電流密度下的長循環性能。
最后,Li@GO-Zn/Cu(預沉積10 mAh cm-2的金屬鋰)作為負極與商業LFP(11.5 mg cm-2)正極以5:1的低N/P比組裝成全電池 (圖6a)。首先,對其軟包電池進行柔性測試,證明即使在受大角度彎曲畸變的影響下,電池仍可穩定地為發光二極管供電(圖6b, c)。這表明制備的柔性電池具有優異的儲鋰性能和機械穩定性,有望在柔性電子領域得到應用。圖6d顯示了LFP||Li@GO-Zn/Cu的電壓平臺在不同的電流密度下波動不大,即使在5C的高倍率電流密度下也相較于其他電池保持較高的比容量和較小的極化電壓(圖6e);當電流密度變回0.1 C時,LFP||Li@GO-Zn/Cu的可逆容量完全恢復,表明基于此形成的全電池具有穩定的可逆性。與其他電極相比,無論電流密度如何變化,在不同充放電平臺上LFP||Li@GO-Zn/Cu始終保持最小極化電壓(圖6f)。同時,長期循環性能測試表明,LFP||Li@GO-Zn/Cu在經歷100周循環后仍有81.1%的容量保持率,與純Cu(0%)、GO/Cu(47.6%)和Zn/Cu(40.6%)相比表現出極高的循環穩定性能(圖6g)。這是因為親鋰的Zn作為成核種子發揮了穩定的作用,促進了初始成核的均勻。具有離子選擇性的GO作為人工保護層有效地錨定了脫合金化的Zn2+,使Zn層在脫鋰過程后仍具備可持續能力,而且高離子電導性的GO亦具有使鋰快速均勻沉積的能力,從而實現高性能鋰金屬電池的構筑。
【結論】
本文通過構建獨特的“三明治”鋰金屬負極結構,系統地研究了氧化石墨烯(GO)在鋰嵌入、脫出過程中對鋅(Zn)成核晶種穩定性的作用。由于氧化石墨烯作為人工SEI層具有較強的Zn2+吸附性和較高的離子電導率,不僅有效阻斷了Zn2+從LiZn合金向電解液的遷移,還能促進鋰從氧化石墨烯表面向Zn層的擴散,從而在Zn層優先沉積。因此,具有離子選擇性的石墨烯能夠保持Zn層的穩定性,協助提高Zn在鋰脫出過程中的穩定性和可逆性。同時,石墨烯界面憑借良好的力學性能,可以克服電池在鋰嵌入、脫出過程中發生的體積變化。因此,GO-Zn/Cu||Li半電池表現出穩定的循環性能,平均CE高達98%,與高載量(11.5 mAh g-1)正極匹配的LFP||Li@GO-Zn/Cu全電池在100周循環后仍表現出高容量保持率(~87%)。本項研究亦發現,在鋰脫出過程中Zn2+很容易從合金中進入電解液,從而影響Zn成核晶種的穩定性。值得注意的是,石墨烯具有的離子選擇性可以保持金屬位點的可持續性。因此,親鋰的鋅層和氧化石墨烯界面層的結合不僅保證了鋰沉積的均勻性,而且維持了成核位點的穩定性,從而提高電池的循環性能。
Jingjing Ma, Jinlong Yang, Can Wu, Meng Huang, Jiawei Zhu, Weihao Zeng, Lun Li, Peng Li, Xin Zhao, Fan Qiao, Zixin Zhang, Daping He*, Shichun Mu*, Stabilizing nucleation seeds in Li metal anode via ion-selective graphene oxide interfaces. Energy Storage Materials, 2023.
https://doi.org/10.1016/j.ensm.2023.01.045