木士春團隊 邃瞳科學云 2024年09月23日 11:07 北京
第一作者:宮蕾
通訊作者:朱加偉博士、木士春教授
通訊單位:武漢理工大學材料復合新技術國家重點實驗室
論文DOI:10.1002/anie.202411125
全文速覽本文以富勒烯(C60)為前驅體,采用堿刻蝕的策略構建了富含五元環缺陷的碳納米材料(PRC),深入探究了五元環缺陷碳的析氫反應(HER)活性。此外,通過碳五元環上的C原子和Ru原子間的p-d軌道雜化效應將活性Ru團簇與PRC耦合(Ru@PRC),進一步提高了催化活性。理論計算和實驗結果都表明電化學性能的改善主要歸因于碳基質中存在的本征五元環缺陷。本征五元環缺陷相對于常規的六元環具有更高的電荷密度和更優化的p帶中心,因此可作為析氫催化活性位點。C原子和Ru原子間的p-d軌道雜化效應有利于誘導反應活性位點的電子重新分布、調節反應中間體的吸附能并促進水解離過程,從而進一步改善電催化活性。
背景介紹由可再生電力驅動的水電解生產綠氫被廣泛認為是實現全球清潔和可持續能源供給的一條很有前途的途徑。開發高效經濟的析氫反應(HER)電催化劑是實現該技術的關鍵之一。憑借著高導電性和耐腐蝕特性,碳基材料成為最具成本效益的候選材料。然而,碳基結構中將會不可避免地存在拓撲缺陷,將會影響其物化性質。然而,拓撲缺陷的本征催化活性和其催化作用機制卻鮮報道。鑒于此,本文采用堿刻蝕策略定向構筑了富本征五元環缺陷的碳納米材料,系統地探究了碳五元環缺陷的本征析氫反應性,并通過五元環中的C原子與活性Ru原子之間的p-d軌道雜化效應進一步提高了材料的電催化活性。本文所揭示的拓撲缺陷催化作用機制為設計和構筑高效、經濟的碳基納米材料電催化劑提供了科學指導。
本文亮點
1. 采用理論計算和實驗驗證相結合的方法,探究了碳五元環拓撲缺陷的本征析氫反應性。
2. 將碳五元環與活性Ru物種耦合后(Ru@PRC)發現,碳五元環上的C原子和Ru原子之間存在的p-d軌道雜化效應,有效調節了Ru的電子態和配位環境,從而優化了HER中間體的結合能,促進了水解離過程。
3. 獲得的Ru@PRC催化劑在全pH范圍內都具有優異的HER活性和穩定性,且其在大電流密度(1000mA cm-2)下的催化活性和穩定性均優于商用Pt/C催化劑,展現出重要的工業應用價值。
圖文解析首先,通過密度泛函理論(DFT)計算,研究了碳五元環的析氫反應性。結果表明,相比于常規的碳六元環,碳五元環具有更低的能帶隙、更優化的p帶中心以及更明顯的局域電子重排,使得碳五元環上的碳原子表現出更高的電荷密度分布,表明碳五元環拓撲缺陷可成為析氫反應的催化活性位點;此外,碳五元環還展示出更低的氫吸附自由能和水解離能壘,有利于吸附氫在活性位點表面的富集和Volmer過程的進行,從而提高了整體的析氫效率。將碳五元環和活性Ru物種耦合(Ru@PRC)后,五元環上的C原子與Ru原子之間的p-d軌道雜化效應促進了局域電子的重排;缺電子Ru位點的d帶中心發生明顯的負移,降低了與氫中間體的結合能,從而進一步提高了復合位點的析氫活性。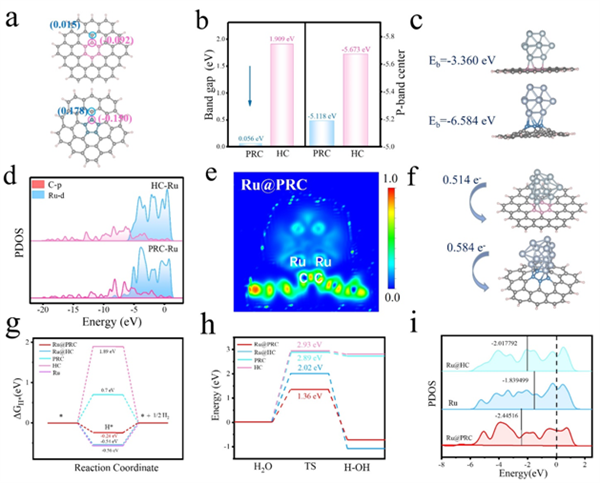
圖1 PRC及Ru@PRC的HER活性起源分析
為了驗證理論計算結果,實驗制備了富五元環缺陷的碳材料。首先選擇具有本征五元環結構的C60作為原材料,對其進行堿刻蝕構建富五元環碳(PRC)。其次,通過濕化學法和熱還原法將活性Ru物種錨定在PRC框架上,獲得了目標產物(Ru@PRC)。再以同樣的合成手段刻蝕石墨烯材料,獲得含有常規六元環碳的材料(HC)和Ru@HC。AC-STEM圖像顯示PRC保留了大量的本征五元環缺陷,引入Ru物種后,碳前驅體的整體形態并未發生明顯改變,Ru團簇(尺寸小于2 nm)均勻地分布在碳基底上。
圖2 PRC及Ru@PRC的合成與結構表征
XRD分析結果表明,堿刻蝕后碳構型發生轉變;Raman、XPS、EPR及同步輻射測試結果都表明富含五元環拓撲缺陷的碳材料具有比常規碳六元環材料更高的缺陷度;Ru的高分辨XPS光譜表明,由于Ru原子和C原子之間的p-d軌道雜化效應,電子由Ru簇向PRC轉移;XAFS分析結果進一步確認Ru@PRC中在2.8 ?處的小峰歸屬于Ru-Ru配位(配位數約為2.2),表明Ru具有典型的團簇特征。
圖3 PRC及Ru@PRC的電子和配位結構調查
進一步考察了PRC及Ru@PRC在寬pH范圍內(0.5 M H2SO4、1 M KOH和1 M PBS)的電催化析氫活性。測試結果表明,PRC在酸、堿、中性介質中均展現出比HC更優異的HER活性,證實了碳五元環對材料電催化活性的貢獻是不容忽視的。當其與活性物種Ru耦合后,Ru@PRC表現出更卓越的電催化HER活性:在堿、酸和中性電解質中,Ru@PRC在10 mA cm?2電流密度下的過電位分別低至28、58和71 mV,均優于含有常規六元環的碳材料(Ru@HC),與理論預測結果一致。此外,Ru@PRC在大電流(1000 mA cm?2)下也具有優異的電催化活性和穩定性,優于商業Pt/C催化劑,展示出很好的工業應用價值。同時,由于Ru金屬價格的低廉,該催化劑還具有出色的質量活性與價格活性,遠高于鉑催化劑。優異的HER性能應歸功于碳五元環拓撲缺陷的存在和Ru原子和C原子之間的p-d軌道雜化效應的協同作用。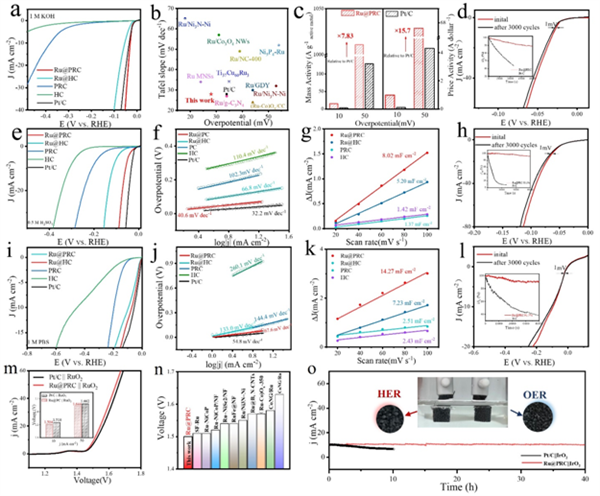
為了更深入地研究拓撲五元環對堿性電化學析氫性能的貢獻,進行了原位Raman光譜測試。圖5a-d中對應于2800~3800 cm?1的拉曼衍射峰分別為HC、PRC、Ru@HC和Ru@PRC在HER過程中的O-H拉伸帶。包絡峰可以分成3220(藍色)、3430(紫色)、3600(橙色)cm?1的三個峰,分別對應4個氫鍵水(4-HBW)、2個氫鍵水(2-HBW)和K+離子水合水(KW)。KW更容易轉化為H-down結構,而H-down結構可以在HER中充當“助催化劑”,持續向界面供水,提高電子傳遞效率,有效加速HER的Volmer過程。Ru@PRC和PRC在任何電壓值下分別都顯示出比Ru@HC和HC更多的KW。這也表明碳五元環拓撲缺陷的存在確實優化了催化劑界面水的吸附構型,從而提高了堿性HER活性。此外,Ru@PRC具有比Ru@HC更高的親水性,表面的親水性可以提高對表面活性位點的利用率,進而加速反應動力學。
圖5 PRC及Ru@PRC的催化機制分析
總結與展望綜上所述,本文設計并構建了富含五元環缺陷的碳納米材料(PRC)。密度泛函理論(DFT)計算表明,由于更高的電荷密度、更優化的p帶中心和更低的能帶隙,碳基質中的本征五元環具有出色的電化學反應性,因此可以作為析氫反應中的潛在活性位點。鑒于本征五元環缺陷對碳納米材料的有利影響,通過刻蝕富勒烯(C60)巧妙地制備了五邊形缺陷富碳納米材料(PRC)。當與Ru耦合(Ru@PRC)后,由于C原子與Ru之間強的p-d軌道雜化效應,使得材料的電化學性能顯著提升。實驗表明,Ru@PRC在全pH范圍內都展現出優異的HER活性和穩定性。該工作提高了對拓撲缺陷在調節碳基電催化劑活性和選擇性方面的基本認識,同時亦為可控構筑本征缺陷碳納米材料提供了一種有效的途徑。
作者及團隊介紹宮蕾:武漢理工大學木士春教授課題組2024級博士研究生,主要研究方向為氫能相關催化劑的設計合成與催化機制研究。
朱加偉:武漢理工大學木士春教授課題組2023屆博士畢業生,目前為香港城市大學博士后,主要研究方向為貴金屬基功能材料的可控構筑、富缺陷碳基催化劑的設計與機制研究。
木士春:武漢理工大學首席教授,博士生導師,國家級高層次人才。長期致力于質子交換膜燃電池、電解水催化劑及鋰離子關鍵材料研究。以第一作者或通訊作者在Nat. Commun.、Adv. Mater.、J. Am. Chem. Soc.、Angew. Chem. Int. Ed.、Energy Environ. Sci.、Nano Lett.等國內外期刊上發表300余篇高質量學術論文。
團隊介紹:武漢理工大學先進能源材料研究團隊依托材料復合新技術國家重點實驗室,長期從事質子交換膜燃料電池關鍵材料與核心器件、電化學產氫催化材料、鋰離子電池電極材料和碳納米材料等研究工作。歡迎有志于科技報國的研究生及博士后加入團隊!
課題組主頁http://m.714744.com/ss/shichunmu/index.html