����˹��ϳɸ߷��Ӳ��ϱ��V����(y��ng)�����������a(ch��n)�ķ������档�߷��ӵľ���(zh��n)���ɿغϳ�ʹ�ò��Ͼ��и��õĿ��{(di��o)���ԣ��Ķ������չ�˸߷��ӵđ�(y��ng)�÷������S���ɿ�/���Ը߷��ӾۺϷ����İl(f��)չ���Ծ��Ը߷��Ӟ�������ϳ����K���Ķ��ӴΏ�(f��)�s�߷����Լ��߷��ӏ�(f��)�ϲ���Ҳ���Ժϳɲ���(y��ng)�á�
�߷��ӵĆ��w�M�ɡ����������������ֲ��ȶ���Ӱ푸߷��Ӳ������ԵĻ������ء�����֮�⣬�߷��ӵ���?f��)�Y(ji��)��(g��u)Ҳͬ����Ҫ����ͬ��?f��)�Y(ji��)��(g��u)�ĸ߷��ӣ������h(hu��n)�֧�������ͣ�ˢ��߷��ӣ�����ھ������������|(zh��)���l(f��)�����^���׃�������У�������֧���߷��ӣ���ճ�ȵ͡��ĩ�˹��ܶȸߡ����Ӄ�(n��i)��϶������c(di��n)���ڝ�(r��n)�����dˎ�ȷ��������V���đ�(y��ng)��ǰ����
Ŀǰ�ϳɣ�����֧���߷��ӵķ�������Ҫ�ЃɷN������ܶȆ��wABf (f��2) �Ŀs�ۣ��Լ�AB* �͆��w���Կs����ϩ���ۺϣ�Fr��chet��Science 1995, 269, 1080-1083������AB*�͆��w�У�A��ɾۺϵ���ϩ����B*�����l(f��)λ�c(di��n)��Ȼ�����@�ɷN�����ڄ�(d��ng)���W(xu��)������ۺϙC(j��)�ƣ�����y�Եõ�խ�������ֲ���֧���߷��ӣ��ҟo(w��)����(sh��)�F(xi��n)��ָ��λ�c(di��n)���x�������l(f��)��
�b�ڿɿ�/���Ծۺ��ھ��Ը߷��Ӻϳ���ȡ�õľ�ɹ���Ү����W(xu��)����������n�}�Mϣ���_(k��i)�l(f��)һ�N��(ji��n)�ģ��m���ڶ�N��Ҋ(ji��n)���w�Ŀɿ�/����֧���ۺϷ��������Դ˷������ض�λ�c(di��n)����(zh��n)����֧���߷��ӡ��ϳ����K���Ķ��γɾ��в�ͬ�Ӵεĸ߷��ӽY(ji��)��(g��u)����D1a�����ڴ�֮ǰ��Yokozawa��Yamago���˾���(b��o)���^(gu��)���á��ۺ��T��(d��o)֧�����Ć��w�O(sh��)Ӌ(j��)˼·����(l��i)��(sh��)�F(xi��n)֧���߷��ӏ��ض�λ�c(di��n)�����L(zh��ng)��Angew. Chem. Int. Ed.2009, 48, 5942-5945; Nat. Commun.2017, 8, 1863.����Ȼ�����w�ķN��^���һ��
��ˣ�����?c��)O(sh��)�����æ�-�u��ϩ�N�����-�����ϩ��������������ϩ����w��ԭ���D(zhu��n)�����ɻ��ۺϣ�ATRP���ėl�����M(j��n)�й��ۣ��Դˌ�(sh��)�F(xi��n)���c(di��n)���l(f��)��ͬ�r(sh��)��(sh��)�F(xi��n)�ɿص��?zh��n)��ۺϣ����،��ˆ��w��ʹ�÷������D1b�������O(sh��)Ӌ(j��)���P(gu��n)�I�c(di��n)���ڦ�-�u��ϩ�N������������ATRP���l(f��)�������ڰl(f��)���ۺϡ����p�I���ӳ�׃?y��u)���I֮��ԭ�Ȧ�λ��̼�u�I������Ķ��ɞ��µ�ATRP���l(f��)λ�c(di��n)��

�D1��a����֧���߷��Ӟ����K�ď�(f��)�s�߷��ӽY(ji��)��(g��u)����b������ATRP��֧���ۺϷ����ķ���(y��ng)�C(j��)��
�ϳɷ������ġ�һ偡��������w�ľ����μ�
���о�����ʼ�A�Σ������á�һ偡�����(l��i)�о���-�����ϩ����������BBA���ͱ�ϩ����������nBA����ATRP�l���µ����ɻ����ۡ����ڶ���������^��BBA��nBA���ĵÿ�һ��(g��)��(sh��)����(j��)���ҡ��@һ���棬��(d��o)�µõ��Ĺ�����ĽM�ɷֲ�����������BBA��֧����λ�c(di��n)�������w�F(xi��n)��֧���߷�����֧���c(di��n)�ֲ��IJ�����
��һ���棬ÿ����һ����BBA���͕�(hu��)����һ��(g��)�µ����l(f��)λ�c(di��n)�����ɻ�����S֮���ߣ����ɻ��p���Kֹ�ĸ���Ҳ����(y��ng)��������ڡ�һ偡����У����BBA��Ͷ�ϱ���ߣ��ۺ����ʕ�(hu��)Ѹ�ٽ������Kֹ����������朽Kֹ����������ע��þ���ע��ڶ����wBBA�ķ����M(j��n)�й��ۣ��Ķ��õ��߷��������ͷ������ֲ������?= 1.2������֧���c(di��n)�ֲ��������֧���߷��ӡ�ͨ�^(gu��)�{(di��o)��(ji��)BBAע��Į�(d��ng)�����ٶȣ������ɹ���(sh��)�F(xi��n)�˲�֧ͬ���ܶȵľ۱�ϩ����������PnBA�����۱�ϩ�������PMA���;۱���ϩ��PSt���ĺϳɡ�
֧���ۺ���ı���
�ںϳ���һϵ�з������ӽ���֧���Ȳ�ͬ��PnBA֮��������(du��)��˴�̼�V��13C-NMR���M(j��n)���˷������D2a�����S��֧���ܶȣ���BBA�ۺ϶ȣ������ӣ�PnBA���ĩ�˹��܈F(tu��n)�Լ�֧��λ�c(di��n)���ļ�(j��)̼�ĺ˴���̖(h��o)��u����(qi��ng)���C��(sh��)��PnBA��֧���Y(ji��)��(g��u)�����ң��˴�̼�V�ϛ](m��i)�к�����ļ�(j��)̼��̖(h��o)���C���ۺϺ��BBA���ИO�ߵ����l(f��)Ч�ʡ�
���⣬����֧���߷��Ӹ��Ӿo�ܵĽY(ji��)��(g��u)�������z�Bɫ�V��GPC�����������@������Փ������������߀����MALLS-GPC�ͺ˴Ō�(du��)�ۺϷ���(y��ng)��(d��ng)���W(xu��)�M(j��n)���˱O(ji��n)�y(c��)�����l(f��)�F(xi��n)�����ڵͷ������^(q��)֮�⣬Mn,MALLS��Mn,theo�dz��Ǻϣ��D2b�����@�f(shu��)�������_(k��i)�l(f��)��֧���ۺϷ���(y��ng)�ǿɿص��?zh��n)��ۺϡ?
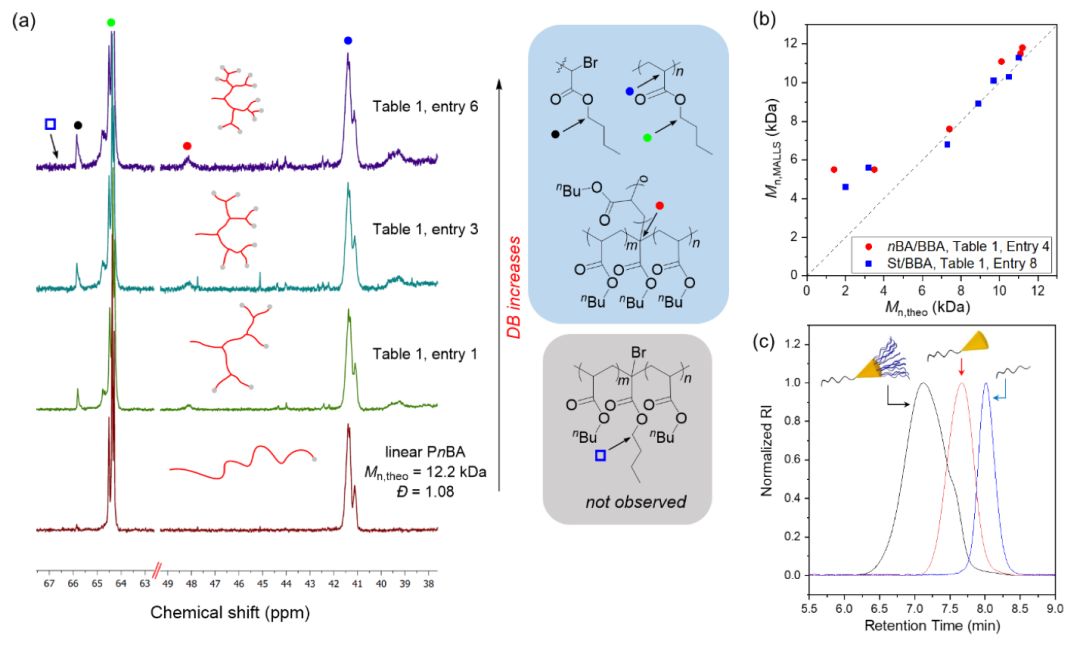
�D2��a�����ԺͲ�֧ͬ���ȵ�PnBA��13C-NMR�V�D����b��֧���ۺϵĽ^��(du��)��������Mn,MALLS��-��Փ��������Mn,theo���D����c�����z�Bɫ�V��GPC������������PDMS66-BP��������l(f��)�����{(l��n)����PDMS66-b-P(nBA90-co-BBA5)���t���Լ�PDMS66-b-P(nBA90-co-BBA5)-b-PSt319���ڣ�
�ģ���λ�c(di��n)�ģ���������l(f��)�����l(f��)�ϳɏ�(f��)�s�ĸ߷��ӽY(ji��)��(g��u)
�������������ò�ͬ�Y(ji��)��(g��u)�Ĵ�������l(f��)������(l��i)�ϳɾ��в�ͬ�Ӵεď�(f��)�s�߷��ӽY(ji��)��(g��u)����D2c��ʾ�����ĩ�������ATRP���l(f��)λ�c(di��n)�ľ۶��������飨PDMS�����l(f��)�������ϳ���PDMS-b-P(nBA-co-BBA)��Ƕ�ι�������M(j��n)һ���U(ku��)朵õ�PDMS-b-P(nBA-co-BBA)-b-PSt��Ƕ�ι����ͬ�ӵģ�����֧����Ԫ�����;ۺ��P�ۺ���ˢҲ�����Դ˷�ʽ�õ����D3����ֵ��һ����ǣ��ڴ�������l(f��)����֧���ۺ��wϵ�У����](m��i)�еͷ������������l(f��)�a(ch��n)�����ɣ��D2c��3b��3d����
�@�C���˴�֧���ۺϷ���(y��ng)���x�������l(f��)�����M(j��n)һ��ӡ�C��������O(sh��)�룬��BBAֻ���ھۺ�֮��̼���I�ű�������⣬�����ϳ���PDMS��֧����PSt��Ƕ�ι��������(du��)���ԽM�b�О��M(j��n)�����о����l(f��)�F(xi��n)���ԽM�b�ߴ�С��ͬ�ȷ������ľ���PDMS-b-PStǶ�ι�����ijߴ磨�D4�����@һ�F(xi��n)���M(j��n)һ���C��(sh��)����֧���Y(ji��)��(g��u)��

�D3���ϳɎ���֧��PnBA��(c��)朵����;ۺ��a���;ۺ���ˢ��c���Լ���(du��)��(y��ng)��GPC����������b���ͣ�d��
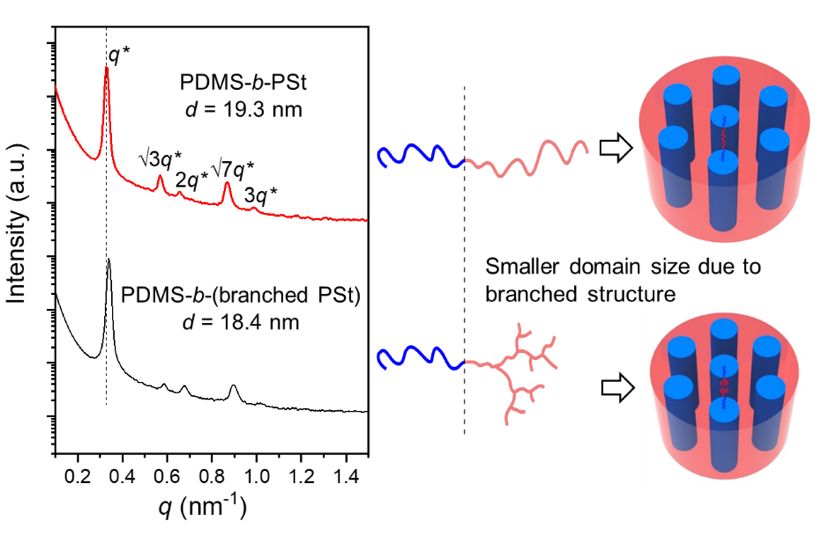
�D4��С��X�侀ɢ�䣨SAXS����(sh��)��(j��)�����ϣ�PDMS66-b-PSt112�����£�PDMS66-b-(PSt112-co-PBBA8)
ͨ�^(gu��)����y(t��ng)��ATRP���������͵�AB*���ۆ��wBBA�������ɹ����_(k��i)�l(f��)�����m���ڶ�N��ϩ�����w�����ܶ��c(di��n)���l(f��)�ĺϳ�֧���߷��ӵĿɿؾۺϷ������߷��ӵ�֧���ȿ���ͨ�^(gu��)�{(di��o)��(ji��)BBA��Ͷ�ϱ��M(j��n)���{(di��o)�ء�ԓ�ۺϷ���ͬ���m���ڶ�λ�c(di��n)�Ĵ�������l(f��)�����Ķ�������(sh��)�F(xi��n)�ˌ�֧���߷��ӡ�ģ�K�����ĺϳ��O(sh��)Ӌ(j��)˼·��Ŀǰ��ֹ�����ô˾ۺϷ����õ���֧���߷����ϵ�ƽ��֧���c(di��n)��(sh��)Ŀ߀���^���ޡ��C�����������֧���ܶȡ��M(j��n)һ���،����w�m�÷�����������չ�������Ļ��Ծۺ��wϵ������ֵ��̽�����о�����Ҳ�dz�ϣ���и���ĸ߷��ӻ��W(xu��)�о����뵽�@һ�о��I(l��ng)���Ё�(l��i)��
ԓ�ĵĵ�һ���ߞ�Ү����W(xu��)���W(xu��)�c�h(hu��n)������ϵ����������n�}�M��ʿ����Ͳ�ʿ���܉�(m��ng)ѩ��������������߀�����T���x�����Ǿ_����Ц����ͨӍ���ߞ������������
Ү����W(xu��)����������n�}�Mʼ����2016�꣬�n�}�M�����ڰl(f��)չ����(zh��n)��Ч�Ļ��W(xu��)�����ϳɹ����ЙC(j��)���߷��Ӳ��ϣ���ͨ�^(gu��)��߶ȽY(ji��)��(g��u)��������ՓӋ(j��)�㌦(du��)���������M(j��n)���J(r��n)֪���A(y��)�y(c��)����(y��u)�����n�}�Mؓ(f��)؟(z��)�������2008�ꮅ�I(y��)�ڱ�����W(xu��)���څ��Ɩ|�����n�}�M��ɱ��Ʈ��I(y��)Փ�IJ��@���W(xu��)�͔�(sh��)�W(xu��)�p�W(xu��)ʿ�W(xu��)λ��2013���ڿ���(n��i)��÷¡��W(xu��)Krzysztof Matyjaszewski��Tomasz Kowalewski����ָ��(d��o)�«@���W(xu��)��ʿ�W(xu��)λ�����M(j��n)����ʡ�����W(xu��)Ժ���W(xu��)ϵJeremiah Johnson�ͻ���ϵBradley Olsen�����n�}�M���²�ʿ���о����F(xi��n)��Ү����W(xu��)���W(xu��)�c�h(hu��n)������ϵ�������ڡ�
- �L(zh��ng)����(y��ng)���������A�о��T�F(tu��n)�(du��) JACS����x�ӽY(ji��)�ϴ���(sh��)�F(xi��n)��(1,3-������h(hu��n))�Ŀɿغϳ� 2026-03-27
- ���ݴ�W(xu��)���S/���Ľ�/�Ώ������ڈF(tu��n)�(du��) ACS Nano���༉(j��)��{�F(tu��n)�ؽM�b�w�ľ���(zh��n)�z�w�ۺ� 2026-03-06
- �L(zh��ng)����(y��ng)����ꐌW(xu��)˼/���@�F(tu��n)�(du��) Angew���������Ч��(y��ng)�{(di��o)�ص��p�����F�������h(hu��n)�������c�h(hu��n)�������ɿع���-�ϳɸ߷������������� 2026-03-05
- ����W(xu��)�㽭�о�Ժ���߷��ӏ�(f��)�ϲ������ġ����������\(ch��ng)ƸӢ�� 2024-09-23
- ������������/������ڈF(tu��n)�(du��) Angew�����ڳ�֧���Y(ji��)��(g��u)��(g��u)���ߏ�(qi��ng)���gˮ���z 2023-09-05
- ���A��W(xu��)�S�Aƽ���ڈF(tu��n)�(du��)��ACS Nano�����(x��)����(n��i)��֧���ۺ� - ��ЧҎ(gu��)�ܰ���(x��)������ˎ�� 2023-06-14
�\(ch��ng)���P(gu��n)ע�߷��ӿƼ�

- ��(gu��)�a(ch��n)���h�Ӱ�������^��PMEC...
- ��ʲô2026����ИI(y��)��(hu��)�h����...
- ��ˎ����Ч������(j��)����PMEC C...
- 2026����ߌ��Ї�(gu��)��(sh��)�(y��n)�Ұl(f��)չ...
- �����I(l��ng)Ʊح300+���h��I(y��)�R��...
- �P(y��ng)���Ж|�����L���Ѓx�����{(l��n)...
- �f(w��n)����Ŀ �ذ��_(k��i)�֣�2025�C(j��)...
- ICIE��(gu��)�H(�V��)Ϳ�Ϲ��I(y��)չ�[...
- �_(k��i)չ�ڼ����ۄ�(sh��)������朄�(d��ng)δ...
- 9������Ҋ(ji��n)���£�AI�(q��)��(d��ng)+��(gu��)�a(ch��n)...
- �P(gu��n)���e�k�ڶ�ʮ�����Ї�(gu��)��(gu��)�H...
- �п�Ժ��(d��ng)�������F(tu��n)�(du��) Adv. ...
- �Ͼ����I(y��)��W(xu��)ë����/��҂�...
- �|�ϴ�W(xu��)�����/�����F(tu��n)�(du��) AF...
- �۳Ǵ�Steven Wang���ڡ��^(gu��)...
- �����ֿƴ��䏩/�ǫI(xi��n)�¡���...
- ���������ƾ��_(d��)���ڡ������i...
- �A�ƴ��T�ض����ڈF(tu��n)�(du��) Macro...
- �����������/���t(y��)������� ...
- ������ꐏ�(qi��ng)���ڈF(tu��n)�(du��) Matter...
- �������մ�/���� AFM���w�S...
- �A������߅�������w�i������...