魯璽麗,蔡偉,趙連城
(哈爾濱工業(yè)大學(xué)材料科學(xué)與工程學(xué)院,黑龍江哈爾濱150001)
Synthesis and characterization of poly(L-lactide)
LU Xi-li, CAI Wei, ZHAO Lian-cheng
(School of Materials Science and Engineering, Harbin Institute of Technology, Harbin 150001, China)
Abstract:Poly(L-lactide)(PLLA) was synthesize by ring opening polymerization of l-lactide. Effects of amount of initiator and the polymerization time on the molecular weight were investigated. Experimental results show that high molecular weight PLLA can be obtained by control of the polymerization conditions. The structure of PLLA was confirmed by the FTIR. The DSC result indicates the glass transition temperature of PLLA is 62℃ and the crystallinity is 42.3%. The analysis of TG illustrated that the thermal decomposition temperature is 299℃.
Key words:lactide;polylactide;ring opening polymerization
摘要:采用丙交酯開環(huán)聚合合成聚L-乳酸(PLLA),研究了引發(fā)劑用量、聚合時間對聚L-乳酸分子量的影響。采用FTIR、DSC、TG分析方法對聚乳酸的結(jié)構(gòu)和熱性能加以表征。研究結(jié)果表明:通過對聚合時間以及引發(fā)劑用量的控制,合成出高分子量聚L-乳酸。FTIR分析結(jié)果證實了聚L-乳酸的結(jié)構(gòu),DSC分析表明合成出的聚L-乳酸玻璃化轉(zhuǎn)變溫度為62℃,結(jié)晶度達到42.3%;TG分析表明聚L-乳酸熱分解溫度為299℃。
關(guān)鍵詞:丙交酯;聚乳酸;開環(huán)聚合
中圖分類號:O631.22 文獻標(biāo)識碼:A
文章編號:1001-9731(2004)增刊-2287-03
1 引言
聚乳酸(PLA)是一種重要的可生物降解高分子材料,因其具有良好的生物相容性和生物可吸收性,已在骨折內(nèi)固定、組織工程支架、醫(yī)療植入、外科縫合線、藥物控釋體系等方面得到了廣泛應(yīng)用[1~4]。由于骨折內(nèi)固定和組織工程支架對材料的強度要求較高,因此只有高分子量的聚乳酸才有實際應(yīng)用價值。高分子量聚乳酸的制備一般采用兩步法[5]:第一步,由乳酸脫水環(huán)化先制得丙交酯單體;第二步,由精制的丙交酯開環(huán)聚合制得相對分子質(zhì)量較高的聚乳酸。目前,因在丙交酯的純度和合成工藝等方面存在差異,導(dǎo)致關(guān)于聚乳酸合成方面一些文獻的報道結(jié)果相差懸殊[6,7],且合成工藝的可重復(fù)性較差。
本工作系統(tǒng)研究了開環(huán)聚合生成聚L-乳酸的合成條件及影響分子量的因素,目的是尋求能夠穩(wěn)定合成出高分子量聚L-乳酸的工藝,通過使用1,4-丁二醇復(fù)合引發(fā)劑,進一步探討了羥基在丙交酯開環(huán)聚合中的作用。同時采用FTIR、DSC、以及TG-FTIR聯(lián)用分析方法對聚乳酸的結(jié)構(gòu)和熱性能加以表征,為后續(xù)的研究工作奠定基礎(chǔ)。
2 實驗
2.1 主要原料
L-乳酸:醫(yī)藥級,Purac公司;辛酸亞錫(Sn(oct)2):分析純,中國醫(yī)藥集團上海化學(xué)試劑公司;1,4-丁二醇:分析純,中國醫(yī)藥集團上海化學(xué)試劑公司;乙酸乙酯:分析純,天津化學(xué)試劑一廠。
2.2 聚L-乳酸的制備
采用Kulkarni方法[5]合成丙交酯,粗丙交酯用乙酸乙酯反復(fù)重結(jié)晶,真空干燥。將新鮮丙交酯與一定比例的Sn(oct)2置于充分干燥的聚合管(自制)中,單體與引發(fā)劑的摩爾比n(M)/n(I)為6000~10000。聚合管在高真空下封管,然后在(130±1)℃的恒溫干燥箱內(nèi)反應(yīng)不同時間。冷卻后打碎聚合管,塊狀聚合物用三氯甲烷溶解,甲醇沉淀,真空干燥至恒重。
2.3 聚乳酸的表征
紅外光譜分析用KBr壓片,采用美國Perkin Elmer公司的Spectrum One型紅外光譜儀測定。相對分子量的測定采用粘度法,以三氯甲烷為溶劑,測試溫度為(25±0.01)℃,溶液濃度為0.012~0.025g/mL。計算公式為[η]=5.45×10-4M0.73[8],其中[η]為特性粘度。DSC分析在法國塞特拉母公司DSC-141型差熱掃描量熱分析儀上進行,氮氣保護(40mL/min),試樣從-50℃以10℃/min升溫速率升溫至200℃,從DSC曲線上得出聚乳酸的玻璃化轉(zhuǎn)變溫度(Tg)和結(jié)晶度。TG-FTIR聯(lián)機分析在美國Perkin Elmer公司的Spectrum One-Pyris1上測定聚乳酸熱分解,以20℃/min的升溫速度從30℃加熱到450℃。
3 試驗結(jié)果與討論
3.1 聚乳酸的合成
3.1.1 聚合條件對聚乳酸分子量的影響
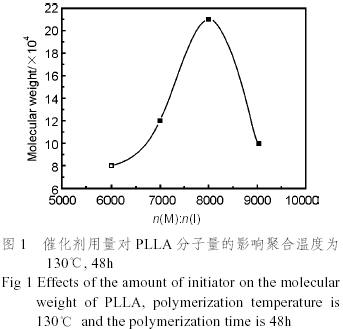
由于無毒和高效,Sn(oct)2常常是丙交酯開環(huán)聚合的首選引發(fā)劑,丙交酯開環(huán)聚合的機理是配位聚合機理。圖1為引發(fā)劑用量對聚L乳酸分子量的影響,聚合條件是在130℃封管聚合48h。從圖中可以看出,當(dāng)單體和引發(fā)劑的摩爾比n(M):n(I)=8000時,聚乳酸的分子量達到最高值為21×104。引發(fā)劑用量過多(n(M):n(I)小),活性點太多,自然每個活性點所能增長的單體數(shù)目減少,聚合物的分子量不高;引發(fā)劑用量過少(n(M):n(I)大),活性點的數(shù)目少,不足以引發(fā)全部單體的聚合,產(chǎn)物的分子量也低,這與文獻報道[9]的規(guī)律是一致的。圖2為聚合時間對聚乳酸分子量的影響,聚合條件為n(M):n(I)=8000:1,130℃封管聚合。從圖中可以看出聚合反應(yīng)時間對聚乳酸的分子量有一定的影響,聚合時間短,聚合反應(yīng)沒有進行完全,所得的聚乳酸分子量較低,聚合反應(yīng)時間過長,所聚合得到的聚乳酸又受熱有一定程度的降解,這種情況在聚合反應(yīng)溫度較高時,更為嚴(yán)重。

3.1.2 辛酸亞錫加1,4-丁二醇復(fù)合引發(fā)劑對聚乳酸分子量的影響
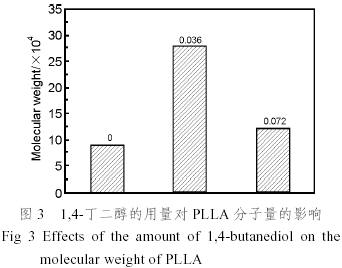
有文獻認(rèn)為[9]丙交酯開環(huán)聚合對羥基的存在非常敏感,羥基是一給電子基團,能與Sn原子發(fā)生強烈配位絡(luò)合作用,使Sn(oct)2失去活性,對反應(yīng)進程產(chǎn)生嚴(yán)重影響;另一方面,羥基參與反應(yīng)的鏈引發(fā)、轉(zhuǎn)移、終止,使反應(yīng)難以得到有效控制和重復(fù),最終導(dǎo)致聚合物分子量大幅度下降。還有研究者[10]認(rèn)為辛酸亞錫只是催化劑,真正的引發(fā)劑是體系內(nèi)的極少量雜質(zhì)(如水或含羥基化合物ROH等)。本文采用帶有兩個羥基的1,4-丁二醇和Sn(oct)2作為復(fù)合引發(fā)劑,初步考察羥基對聚合反應(yīng)的影響。由圖3可見,少量1,4-丁二醇的存在,使聚乳酸的分子量有較大程度的增加,粘均分子量達到28×104,而當(dāng)其含量略有增加時,聚合物的分子量又下降較大,說明聚合反應(yīng)對羥基非常敏感,極少量羥基的存在對丙交酯開環(huán)聚合反應(yīng)有利,有利于合成出高分子量的聚乳酸。
3.2 聚乳酸的表征
3.2.1 聚乳酸的結(jié)構(gòu)表征
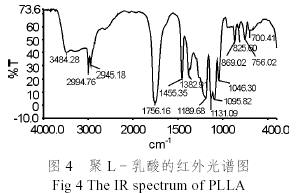
圖4為聚乳酸的紅外光譜圖。從圖4中可以看出,在1756cm-1的吸收峰屬C=O的伸展振動,1189 cm-1峰和1131 cm-1屬C-O伸展振動,證明存在酯基團,2900 ~3000 cm-1之間的峰當(dāng)屬C-H的伸展振動,1455 cm-1峰屬C-H的變形振動,1382 cm-1峰屬甲基的特征峰,證實了聚乳酸的結(jié)構(gòu)。
3.2.2 聚乳酸的熱性能
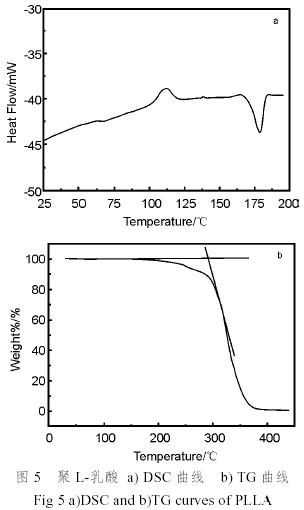
圖5a)和b)分別示出了聚乳酸的DSC曲線和TG曲線。由DSC分析可知,聚乳酸的玻璃化轉(zhuǎn)變溫度為62℃,結(jié)晶熔融熱為39.42J/g,從結(jié)晶熔融熱計算試樣的結(jié)晶度(Xc%)為42.3%,完全結(jié)晶的PLLA的結(jié)晶熔融熱取93.1J/g[10]。從聚乳酸的熱重分析譜可以看出,聚乳酸的熱分解溫度為299℃。
4 結(jié)論
(1)采用辛酸亞錫為引發(fā)劑,通過對引發(fā)劑用量和聚合時間的控制,合成出粘均分子量為21×104的高分子量聚L-乳酸。
(2)采用極少量的1,4-丁二醇作為復(fù)合引發(fā)劑可顯著提高聚L-乳酸的分子量。
(3)紅外光譜證實了聚乳酸的結(jié)果,DSC結(jié)果表明合成出聚乳酸的玻璃化轉(zhuǎn)變溫度為62℃,結(jié)晶度為42.3%。TG分析表明聚乳酸的熱分解溫度為299℃。
參考文獻:
[1] Kricheldorf H R. [J]. Syntheses and Application of Polylactides. Chemosphere. 2000,43:49-54.
[2] Middleton J C, Tipton A J. Synthetic Biodegradable Polymers as Orthopedic Devices.[J]. Biomaterials, 2000, 21:2335-2346.
[3] Kataoka K, Harada A, Nagasaki Y. Block copolymer micelles for drug delivery: design, characterization and biological significance.[J]. Advanced Drug Delivery Reviews, 2001,47:113-131.
[4] 吳之中, 張政樸, 魯格,等. 聚乳酸的合成降解及在骨折內(nèi)固定材料的應(yīng)用.[J]. 高分子通報, 2000, 3:73-79.
[5] Kulkarni B K, Moore E G, Hegyeli A F, et al. Biodegradable Poly(lactic acid) Polymers.[J]. J BIomed MaterRes, 1971,5:169-181.
[6] Schwach G, Coudane J, Engel R, et al. More about the Polymerization of Lactides in the Presence of Stannous Octoate. [J]. Journal of Polymer Science: Part A: Polymer Chemistry. 1997,35:3431-3440.
[7] Hyon S H, Jamshidi K, Ikada Y. Synthesis of Polylactides with Different Molecular Weights.[J]. Biomaterials, 1997,18:1503-1508.
[8] Schindler A, Harper D.[J]. J Polym Sci Polym Chem, 1979, 17: 2593.
[9] 盧澤儉, 廖凱榮, 李洪權(quán),等. 高強度聚(L-乳酸)骨折內(nèi)固定器件研制Ⅰ.高相對分子質(zhì)量聚(L-乳酸)的合成.[J]. 中山大學(xué)學(xué)報, 1999, 38: 36-39.
[10] Kricheldorf H R, Kreiser-Saunders I, Caroline Boettcher. Polylactones: 31. Sn(Ⅱ)octoate-Initiated Polymerization of L-Lactide: A Mechanistic Study.[J]. Polymer, 1995, 36: 1253-1259.
作者簡介:魯璽麗(1975-),女,吉林渾江人,在讀博士,從事醫(yī)學(xué)生物材料的研究。
(E-mail:luxili@hit.edu.cn),Tel:0451-86412163
論文來源:中國功能材料及其應(yīng)用學(xué)術(shù)會議,2004年,9月12-16日
